Курсовая работа: Тонкослойная хроматография и ее роль в контроле качества пищевых продуктов
Курсовая работа: Тонкослойная хроматография и ее роль в контроле качества пищевых продуктов
КУРСОВАЯ РАБОТА
тонкослойная хроматография
и ее роль в контроле качества пищевых продуктов
Содержание
Введение
Глава 1. Физико-химические основы тонкослойной
хроматографии
1.1 Классификация хроматографических методов анализа
1.2 Основы метода ТСХ
1.3 Распределительная хроматография на бумаге
1.4 Тонкослойная хроматография
1.4.1 Сорбенты
1.4.2 Растворители
Подбор подвижной фазы (системы) проводится по следующим
правилам:
1.4.3 Подготовка пластин
1.4.4 Техника нанесения исследуемых растворов
1.4.4 Хроматографирование
1.4.5 Восходящая тонкослойная хроматография
1.4.6 Нисходящая тонкослойная хроматография
1.4.7 Горизонтальная тонкослойная хроматография
1.4.8 Радиальная тонкослойная хроматография
1.4.9 Сушка пластин
1.4.10 Идентификация разделенных веществ
1.4.11 Физические методы
1.4.12 Значение Rf
1.4.13 Цветные реакции
1.4.14 Сравнение со свидетелем
1.4.15 Физико-химические методы идентификации
1.4.16 Методы количественного анализа
1.4.17 Способы проведения ТСХ
Глава 2. Контроль качества пищевых продуктов посредством
метода ТСХ
2.1 Определение ДДТ, ДДЭ, ДДД, альдрина, дильдрина,
гептахлора, кельтана, метоксихлора, эфирсульфоната и других ядохимикатов в
продуктах питания хроматографией в тонком слое
2.2 Определение
о,о-диэтил-8-(6-хлорбензоксазолинил-3-метил)-дитиофосфата (фозалона) в яблоках
методом тонкослойной хроматографии
Глава 3. ТСХ – оборудование
3.1 Оборудование фирмы НТЦ ЛЕНХРОМ для инструментальной ТСХ
3.1.1 Портативный набор для ТСХ
3.1.2 Базовый набор для ТСХ
3.1.3 Оборудование для проведения ВЭТСХ анализов
3.1.4 Пластины многократного пользования для
высокоэффективной и аналитической ТСХ
3.1.5 Контроль эффективности разделения
3.1.6 Хроматографические камеры
3.1.7 Постхроматографическая дериватизация
3.1.8 Оборудование для нанесения образцов
3.1.9 Приборы и оборудование для высокоэффективной
тонкослойной хроматографии
Литература
Введение
Хроматография, обязательно включающая процесс разделения смесей веществ в динамическом режиме, охватывает не только достаточно обширный раздел аналитической химии, но и лежит в основе ряда технологических процессов. В связи с этим, хроматография включает два основных направления: информационное и технологическое. Первое обеспечивает информацию о качественном и количественном составе и физико-химических свойствах исследуемых объектов, второе — получение материальных продуктов.
Как способ анализа хроматография является частью той группы методов, которая ввиду сложности исследуемых объектов включает предварительное разделение исходной сложной смеси на относительно простые, например, дистилляцией, зонной плавкой, экстракцией, диффузией или комбинацией этих методов. Среди них хроматографические методы обладают наиболее эффективными разделительными возможностями за счет использования большого числа типов межмолекулярных взаимодействий. Стадия разделения в хроматографической колонке или слое сорбента обеспечивает получение относительно простых смесей, анализируемых затем обычными химическим, физико-химическим или физическим методами анализа или специально созданными для хроматографии методами или приемами.
Тонкослойная хроматография (ТСХ, TLC) - один из наиболее используемых методов хроматографического анализа, но наименее популяризируемый.
Несмотря на существовавшие до недавнего времени существенные недостатки, она широко используется для качественного анализа смесей, в основном, за счет дешевизны и скорости получения результатов. [1]
Тонкослойная хроматография имеет множество возможностей и преимуществ, и может быть не только качественным методом анализа. И в то же время это – метод, требующий определенные навыки и знания, без которых он не может существовать.
Глава 1. Физико-химические основы тонкослойной хроматографии
1.1 Классификация хроматографических методов анализа
Разнообразные варианты хроматографии [1] укладываются в относительно простую схему классификации в зависимости от используемой подвижной фазы и характера межмолекулярных взаимодействий. Поскольку характер взаимодействий может быть очень различным – от чисто ситового эффекта к физической сорбции и далее к хемосорбции, то почти не существует объектов, для разделения которых не удавалось бы найти подходящего сорбента и систем растворителей. Области применения основных вариантов хроматографии в зависимости от молекулярной массы исследуемых соединений показаны на рис. 1.
В области молекулярного анализа органических соединений хроматография преобладает над другими методами разделения, не заменяя их.
Классификация вариантов хроматографии приведена в таблице 1 и на рис. 2. Следует иметь в виду, что в аналитической практике преобладает использование варианта проявительной хроматографии, когда подвижная фаза подается в хроматографическое устройство непрерывно, а разделяемая проба — периодически.
При всем разнообразии вариантов хроматографии практически всегда реализуется общая схема процесса, представленная на рис. 3. Подвижная фаза (газ-носитель или жидкость) непрерывно пропускается через слой гранулированного сорбента, засыпанного в колонку.
В этот поток дозирующим устройством вводится импульсно анализируемая смесь, которая должна быть газообразной или испаряться в дозаторе в случае газовой хроматографии, или растворяться в подвижной фазе в случае жидкостной. Перемещаясь потоком подвижной фазы по колонке, анализируемая смесь разделяется на составляющие ее компоненты: компоненты, сорбирующиеся хуже на данном сорбенте, двигаются быстрее и вымываются из колонки раньше, чем сорбирующиеся лучше.
Расположенный после колонки детектор фиксирует наличие в потоке компонентов; его сигнал, обычно пропорциональный концентрации или количеству компонента, записывается на самопишущем потенциометре (регистраторе) в виде хроматограммы — графика зависимости концентрации (количества от времени). Хроматограмма при полном разделении компонентов состоит из системы колоколобразных кривых, называемых пиками: каждый пик относится к одному или нескольким компонентам и соответствует возрастанию, а затем снижению концентрации в потоке подвижной фазы.
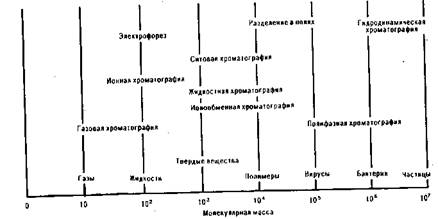
Рис. 1. Области применения
основных вариантов хроматографии в зависимости от молекулярной массы
исследуемого вещества
Таблица 1. Варианты хроматографии по фазовым состояниям
|
Подвижная фаза |
Неподвижная фаза |
Название варианта | |
| частное | общее | ||
| Газ | Адсорбент | Газоадсорбционная | Газовая хроматография |
| Жидкость | Газожидкостная | ||
| Жидкость | Адсорбент | Жидкостно-адсорбционная | Жидкостная хроматография |
| Жидкость | Жидкостно-адсорбционная | ||
| Газ или пар в сверхкритическом состоянии | Адсорбент | Флюидно-адсорбционная | Флюидная хроматография |
| Жидкость | Флюидно-жидкостная | ||
|
Коллоидная система |
Сложная композиция твердых и жидких компонентов | Полифазная хроматография | |
Кроме рассмотренной колоночной, имеются различные варианты так называемой планарной хроматографии, при которых слой сорбента создается не в колонке, а на плоской поверхности, например, как в тонкослойной или бумажной хроматографии.
Область использования газовой хроматографии – соединения с молекулярной массой порядка до 500, которые составляют ~ 5% от общего числа известных соединений. Соединения с большой молекулярной массой — сфера приложения жидкостной хроматографии — составляют 95% всех известных химических веществ. Вместе с тем не следует забывать, что упомянутые 5 % более простых веществ – объектов газовой хроматографии сегодня да и в будущем,— составляют 70 – 80% соединений, которые использует человек в сфере производства и быта. Тот метод хорош, который позволяет решить ту или иную задачу с нужной точностью и достоверностью, экспрессностью и минимальными затратами. [1]
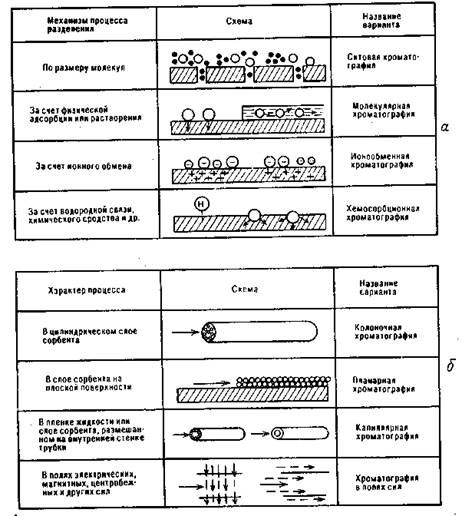
Рис. 2. Варианты хроматографии по характеру взаимодействий (а)
и по способу проведения (б)
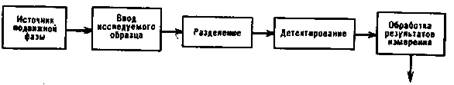
Рис. 3. Схема реализации хроматографического процесса с целью анализа смесей
1.2 Основы метода ТСХ
Основой тонкослойной хроматографии является адсорбционный метод, хотя также встречается метод распределительной хроматографии. Адсорбционный метод основан на различии степени сорбции-десорбции разделяемых компонентов на неподвижной фазе. Адсорбция осуществляется за счет ван-дер-ваальсовских сил, являющейся основой физической адсорбции, полимолекулярной (образование нескольких слоев адсорбата на поверхности адсорбента) и хемосорбцией (химического взаимодействия адсорбента и адсорбата).
Для эффективных процессов сорбции-десорбции необходима большая площадь, что предъявляет определенные требования к адсорбенту. При большой поверхности разделения фаз происходит быстрое установление равновесия между фазами компонентов смеси и эффективное разделение. Так физическое выражение адсорбции-десорбции в упрощенном виде можно выразить уравнением [2]
Г=(Г~/К)с.
где Г~ - предельно возможная величина адсорбции, К – константа равновесия;
с –концентрация абсорбата.
В более строгих подходах к теории адсорбции необходимо учитывать взаимодействие между адсорбированными частицами, неоднородность поверхности, давление, температуру и т.д.
Но как видно из вышеописанного уравнения адсорбция является линейной функцией концентрации.
Еще одним видом используемом в методе тонкослойной хроматографии является распределительная жидкостная хроматография.
В распределительной хроматографии обе фазы - подвижная и неподвижная - жидкости, не смешивающиеся друг с другом. Разделение веществ основано на различии в их коэффициентах распределения между этими фазами.
Впервые метод тонкослойной хроматографии заявил о себе как "Бумажная тонкослойная хроматография", которая основывалась на распределительном методе разделения компонентов. [3]
1.3 Распределительная хроматография на бумаге
В связи с тем, что используемая в этом методе хроматографическая бумага (специальные сорта фильтровальной бумаги) содержат в порах воду (20-22%), в качестве другой фазы используются органические растворители. Использование хроматографии на бумаге имеет ряд существенных недостатков: зависимость процесса разделения от состава и свойств бумаги, изменение содержания воды в порах бумаги при изменении условий хранения, очень низкая скорость хроматографирования (до нескольких суток), низкая воспроизводимость результатов. Эти недостатки серьезно влияют на распространение хроматографии на бумаге как хроматографического метода.
Поэтому можно считать закономерным появление хроматографии в тонком слое сорбента – тонкослойной хроматографии. [3]
1.4 Тонкослойная хроматография
В этом методе хроматографирование веществ происходит в тонком слое сорбента, нанесенного на твердую плоскую подложку. Разделение в этом методе в основном происходит на основе сорбции-десорбции. Использование различных сорбентов, позволило значительно расширить и улучшить этот метод.
В начале появления метода пластины приходилось изготавливать самостоятельно. Но на сегодняшний день в основном используются пластины заводского изготовления, имеющие достаточно широкий ассортимент как по размерам и носителям, так и по подложкам.
Современная хроматографическая пластинка представляет собой основу из стекла, алюминия или полимера (например политерефталат). В связи с тем, что стеклянная основа становится менее популярной (часто бьется, нельзя разделить пластинку на несколько частей не повредив слой сорбента, тяжелая по весу), наибольшее распространение получили пластины, в качестве основ которых используют алюминиевую фольгу или полимеры.
Для закрепления сорбента применяют гипс, крахмал, силиказоль и др., которые удерживают зерна сорбента на подложке. Толщина слоя может быть различна (100 и более мкм), но самый важный критерий - слой должен быть равномерный по толщине в любом месте хроматографической пластинки. [3]
1.4.1 Сорбенты
Наиболее распространенным сорбентом является силикагель.
Силикагель - гидратированная кремниевая кислота, образующаяся при действии минеральных кислот на силикат натрия и сушкой образовавшегося золя. После размалывания золя используют фракцию определенной зернистости (указанную на пластинке, обычно 5-20 мкм).
Силикагель является полярным сорбентом, у которого в качестве активных центров служит группы -ОН. Он легко сорбирует на поверхности воду и образует водородные связи.
Окись алюминия. Окись алюминия является слабо основным адсорбентом и используется в основном для разделения соединений слабоосновного и нейтрального характера. Недостатком пластин на окиси алюминия является обязательная активация поверхности перед использованием в сушильном шкафу при высокой температуре (100-150 0С) и низкая, по сравнению с силикагелем адсорбционная емкость слоя.
Кизельгур - адсорбент, полученный из природных минералов: диатомовых земель. Сорбент обладает гидрофильными свойствами, но более низкой адсорбционной емкостью слоя по сравнению с силикагелем. Кремнекислый магний менее полярный чем силикагель и обычно используется в случаях, когда более полярные адсорбенты не дали эффективного разделения.
Целлюлоза - тонкослойные пластины с нанесенной целлюлозой очень эффективны для разделения сложных органических молекул. Адсорбент представляет собой в основном шарики целлюлозы диаметром до50 мкм, закрепленные на носителе крахмалом. Но, как и в бумажной хроматографии, подъем фронта растворителя происходит очень медленно.
В ионообменных хроматографических пластинках в качестве адсорбента используют ионообменные смолы, содержащие четвертичный аммоний или активные сульфогруппы, участвующие в ионном обмене. Тонкослойная хроматография с такого типа пластинками, проводится с подвижными фазами содержащими сильные кислоты или щелочи. Данные пластинки эффективны для разделения высокомолекулярных и амфотерных соединений.
Вышеперечисленные сорбенты являются наиболее распространенными, но помимо этих существуют множество веществ, используемых как сорбенты. Это тальк, сульфат кальция, крахмал и т.д..
В то же время даже уже указанные сорбенты могут быть модифицированы для придания им новых сорбционных свойств (пропитка сорбентов реактивами, например AgNO3, создание пластин с обращенной фазой). Именно такое разнообразие возможных фаз при минимальных затратах позволяют использовать ТСХ для хроматографирования огромного числа веществ. [4]
1.4.2 Растворители
В тонкослойной хроматографии, в качестве подвижной фазы используют либо чистые вещества (этилацетат, бензол и т.п.), либо смеси веществ (системы) в определенном соотношении.
Подбор подвижной фазы (системы) проводится по следующим правилам:
· Выбирают такую систему, в которой разделяемые компоненты имеют небольшую растворимость (если растворимость вещества высокая, то вещества будут перемещаться с фронтом, при низкой растворимости - оставаться на старте). При распределительной хроматографии или при использовании обращенных фаз, растворимость веществ должна быть выше в подвижной фазе, чем в неподвижной.
· Состав системы должен быть постоянным и легко воспроизводимым.
· Растворитель или компоненты системы не должны быть ядовитыми или дефицитными.
· Система должна полностью разделять вещества близкого строения, причем различия в Rf должно быть не менее 0,05.
· Система не должна вызывать химические изменения разделяемых компонентов.
· В выбранной системе анализируемые вещества должны иметь различные значения Rf и распределяться по всей длине хроматограммы. Желательно, чтобы значения Rf лежало в пределах 0,05-0,85.
· При выборе системы также необходимо учитывать природу разделяемых веществ. Так, при хроматографировании веществ, имеющих основные свойства система не должна обладать кислотными свойствами и наоборот.
Эти рекомендации дают предварительную оценку выбранной системы. Последнее слово все равно остается за экспериментом. [3]
1.4.3 Подготовка пластин
При использовании приобретенных пластин, для хроматографирования их необходимо предварительно подготовить. Это связано с тем, что адсорбенты пластин при хранении сорбируют не только влагу, но и другие вещества, содержащиеся в воздухе. При использовании неподготовленных пластин в процессе хроматографирования появляется фронт "грязи", который может мешать определению веществ, имеющие большие значения Rf, а некоторые вещества, например вода, может изменять состав подвижной фазы, изменяя тем самым получаемые значения Rf.
Предварительная подготовка пластин заключается в разгонке пластин чистым растворителем на всю высоту пластинки (метанол, бензол, диэтиловый эфир),с последующей сушкой пластины в сушильном шкафу при температуре 110-120 0С в течении 0,5-1 часа. Таким способом можно подготовить сразу несколько пластин и при хранении их в сухом герметичном месте, сохраняют свои свойства несколько месяцев. [3, 4]
1.4.4 Техника нанесения исследуемых растворов
Как оказывается, нанесение исследуемого вещества не такая сложная операция, но вместе с тем, она очень влияет на получаемые результаты хроматографирования.
Часто, исследованию подвергаются либо жидкие анализируемые вещества, либо растворы твердых веществ, без какой-либо предварительной пробоподготовки.
Поэтому необходимо всегда помнить ряд моментов, серьезно влияющие на результаты разделения.
Наиболее важным является концентрация наносимых веществ. В ТСХ принято наносить концентрации растворов около 1%. Но с другой стороны чувствительность метода позволяет определять вещества с гораздо меньшими концентрациями.
Если в исследуемом веществе неизвестна общая концентрация компонентов, или известна концентрация, но такого типа вещества еще не хроматографировали, нужно определить какое количество исследуемого раствора достаточно для качественной хроматографии. Существуют несколько приемов, позволяющие это определить.
Для начала нужно нанести несколько пятен хроматографируемых растворов, равные по размеру, но с различным количеством (например 1, 2, 5 мкл) и после хроматографирования изучить форму и размеры разделенных пятен.
Так при правильно подобранной концентрации форма разделенных веществ такая же, как и форма нанесенной на линии старта. Если разделенные пятна имеют большие размеры, чем пятно на старте, то нанесенная концентрация слишком велика. Появление "хвостов", неправильная форма разделенных пятен на пластинке тоже может говорить о высокой концентрации, но может быть вызвана неправильно подобранной хроматографической системой, либо химическим взаимодействием разделяемых компонентов. Подбором количества нанесенного вещества и системы растворителей можно добиться полного разделения на одной пластинке до десяти компонентов в исследуемых веществах. Удобно наносить образцы на специальном столике с трафаретами и подогревом. Нанесение пятен проводят на "линии старта" 1-2 см от нижнего края пластинки. Это необходимо для того, чтобы при опускании пластинки в систему не происходило растворение в ней образцов, а все нанесенное вещество подверглось хроматографированию.
Нанесение растворов проводят либо микрошприцом, либо отградуированными капиллярами. Размер наносимого пятна не должен превышать 4 мм. Это связано с тем, что при большем размере пятна, происходит изменение формы под действием физических сил, да и границы разделенных компонентов могут перекрываться.
Нанесение на пластины исследуемых веществ не должны сопровождаться разрушением сорбента (что довольно сильно влияет на качество разделения), поэтому капля должна наноситься касанием иглы или капилляра о слой сорбента, а не надавливанием. На размер образующегося пятна влияет не только количества наносимого раствора, но и от полярности растворителя и его температуры кипения. Так при нанесении одного и того же вещества в различных растворителях, образовавшееся пятно в котором в качестве растворителя использовался метанол будет больше, чем пятно от раствора хлороформа. С другой стороны при подогреве подложки испарение растворителей будет интенсивнее и размер пятна также уменьшается.
Конечно, проще использовать при нанесении для подсушивания пятен фен, но только в том случае, когда есть полная уверенность, что наносимые вещества не будут окисляться под действием горячего воздуха. Расстояние между наносимыми пятнами должно быть около 2 см.
Иногда при хроматографировании на пластинках наблюдается краевой эффект, в результате чего пятна располагаются не на одной линии а имеют вид подковы, либо по диагонали. Для устранения этого эффекта каждое пятно можно "снабдить" своей дорожкой, отделив нанесенный образец от других путем удаления линии сорбента. Это лучше всего делать под линейку острым предметом (типа скальпеля) но осторожно, чтобы не удалить слишком много сорбента.
После нанесения исследуемых веществ на пластинку, необходимо добиться полного удаления растворителей, так как даже небольшое содержание растворителя в исследуемом веществе может повлиять на разделение и даже изменить состав хроматографической системы.
Удаление растворителей обычно проводят естественной сушкой пластин 5-10 мин, либо при нагревании феном или в сушильном шкафу. [3, 4]
1.4.4 Хроматографирование
Тонкослойная хроматография имеет несколько способов, связанных, в основном, с видом движения растворителей.
· Восходящая тонкослойная хроматография
· Нисходящая тонкослойная хроматография
· Горизонтальная тонкослойная хроматография
· Радиальная тонкослойная хроматография.
1.4.5 Восходящая тонкослойная хроматография
Этот вид хроматографии наиболее распространен и основан на том, что фронт хроматографической системы поднимается по пластинке под действием капиллярных сил, т.е. фронт хроматографической системы движется снизу-вверх. Для этого метода используется наиболее простое оборудование, так как в качестве хроматографической камеры можно использовать любую емкость с плоским дном и плотно закрывающейся крышкой, в которую свободно помещается хроматографическая пластинка.
Метод восходящей тонкослойной хроматографии имеет ряд своих недостатков. Например, скорость поднятия фронта по пластинке происходит неравномерно, т.е. в нижней части она самая высокая, а по мере поднятия фронта уменьшается. Это связано с тем, что в верхней части камеры насыщенность парами растворителя меньше, поэтому растворитель с хроматографической пластинки испаряется интенсивнее, следовательно уменьшается его концентрация и скорость движения замедляется. Для устранения этого недостатка по стенкам хроматографической камеры прикрепляют полоски фильтровальной бумаги, по которым поднимающаяся хроматографическая система насыщает парами камеру по всему объему.
Некоторые хроматографические камеры имеют на дне разделение на две ванночки. Это усовершенствование позволяет не только уменьшить расход хроматографической системы (для получения необходимой высоты хроматогратографической системы требуется меньший объем) но и использовать дополнительную кювету для растворителя, увеличивающего давления насыщенных паров в камере.
Недостатком также можно считать необходимость следить за фронтом растворителя, так как возможно "убегание" лини фронта растворителя до верхнего края. В таком случае определить действительное значение Rf уже не представляется возможным. [3]
1.4.6 Нисходящая тонкослойная хроматография
Этот метод хроматографии основан на том, что фронт хроматографической системы опускается по пластинке в основном под действием сил тяжести, т.е. фронт подвижной фазы движется сверху вниз.
Для этого метода в верхней части хроматографической камеры крепится кювета с хроматографической системой из которой с помощью фитиля на хроматографическую пластинку поступает растворитель, который стекает и происходит хроматографирование исследуемого образца.
К недостаткам этого метода можно отнести усложнение оборудования. Этот метод используется в основном в бумажной хроматографии. [3]
1.4.7 Горизонтальная тонкослойная хроматография
Этот метод наиболее сложен в аппаратурном оформлении но наиболее удобен. Так, в хроматографической камере пластинка размещается горизонтально и подача системы происходит на один край пластинки с помощью фитиля. Фронт растворителя движется в противоположную сторону.
Есть еще один прием, позволяющий предельно упростить камеру. Для этого хроматографическую пластинку на алюминиевой основе слегка изгибают и помещают в камеру. В данном случае система будет поступать с двух сторон одновременно. Для этой цели подходят только пластины с алюминиевой подложкой, так как пластиковая и стеклянная основа "несгибаема", т.е. не сохраняет форму.
К достоинствам этого метода можно отнести то, что в горизонтальной кювете насыщение парами системы происходит гораздо быстрее, скорость движения фронта постоянная. А при хроматографировании с двух сторон, фронт не "убегает". [3]
1.4.8 Радиальная тонкослойная хроматография
Радиальная тонкослойная хроматография заключается в том, что в центр пластинки наносится исследуемое вещество и туда же подается система, которая движется от центра к краю пластинки.
Здесь приведены основные способы хроматографирования в тонкослойной хроматографии. Существует еще достаточно много способов хроматографирования, о которых мы поговорим в дальнейшем.
1.4.9 Сушка пластин
После процесса разделения исследуемых веществ, пластинки сушат. Это тоже немаловажный процесс, так как при наличии на пластинке даже следов растворителя, возможно получить неправильные результаты хроматографирования.
Если хроматографическая система имела в своем составе только легкокипящие компоненты, то достаточно естественной сушки в течении 3-5 минут. Если же в состав системы входят высококипящие жидкости (спирты, вода, органические кислоты и т.д.), сушку пластин нужно проводить не менее 10 мин или помещать пластинку в сушильный шкаф. [3, 4]
1.4.10 Идентификация разделенных веществ
Высушенная пластинка является хроматограммой исследуемых веществ. Если вещества являются окрашенными, то идентификация начинается с определения цвета разделенных веществ.
Но в большинстве случаев разделяемые вещества бесцветны и простое визуальное сравнение невозможно.
Для тонкослойной хроматографии существует несколько видов качественного анализа (идентификации) разделенных веществ:
· Визуальные методы и определение Rf разделенных веществ.
· Цветные реакции.
· Сравнение со свидетелями.
· Физико-химические методы идентификации.
Рассмотрим подробнее каждый вид качественного анализа в тонкослойной хроматографии. [3, 4]
1.4.11 Физические методы
Визуальные методы используются в основном, для определения местоположения пятен разделенных веществ на хроматографической пластинке. Для этого пластинку рассматривают как в видимом свете, так и используя ультрафиолетовый свет (в основном свет с длиной волны 366 и 254 нм). Это первый этап идентификации, на котором определяется качество подобранных условий и полученных результатов хроматографирования.
Так, определив качество хроматографирования (отсутствие "хвостов" разделяемых веществ или перекрытие их пятен, правильную форму и размеры, отсутствие слияния хроматографических дорожек и т.д.) и признании пригодным проведенного разделения для дальнейшего исследования, определяют Rf выявленных пятен. [3]
1.4.12 Значение Rf
Одним из основных показателей в ТСХ является показатель Rf. Этот параметр является аналогией времени удерживания и зависит как от свойств разделяемых веществ, состава подвижной фазы и сорбента, так и от физических параметров. Определение значения Rf проводят как отношение расстояния прошедшего веществом к расстоянию, прошедшего фронтом растворителя
Rf = L/L0
Значение Rf - величина безразмерная и имеет значение от 0 до 1. Однако в литературе нередко встречается такие показатели как hRf, RfЧ100, которые являются тем же Rf, но умноженными на 100, для того, чтобы не оперировать десятичными значениями.
На значение Rf не влияет расстояние пройденное фронтом растворителя, однако во многих методиках описывается прохождение фронта на расстояние 10 см. Это используется только для облечения расчетов Rf.
На практике, в начале определяют расстояние прошедшее фронтом растворителя: от линии старта (а не от края пластинки) до места, где находился фронт в момент окончания хроматографирования. Затем определяют расстояние от линии старта до пятна разделенного вещества. Во тут и оказывает влияние размер пятна! Ведь если пятно имеет круглую форму и небольшой размер, то полученное Rf имеет четкое значение. А если полученное пятно имеет большой размер или неправильную форму, то при определении Rf такого пятна, ошибка может достигнуть 0,1!
В случае распределительной хроматографии коэффициент распределения вещества и его Rf связано соотношением:
![]()
где Sп и Sн - площади поперечных сечений подвижной и неподвижной фазы.
Как мы видим, коэффициент распределения, при постоянном отношении Sп/Sн есть величина пропорционально зависящая от Rf , и может быть определена через него. [3]
1.4.13 Цветные реакции
Цветные реакции в тонкослойной хроматографии используются чрезвычайно широко. Они служат не только для определения местоположения разделенных компонентов (обработка серной кислотой, парами йода), но и определения как класса веществ, так и идентификации (при наличии индивидуальных реакций). При совпадении всех качественных реакций и совпадении полученных значений Rf вещества в трех различных системах с литературными данными, вещество идентифицировано. [3]
1.4.14 Сравнение со свидетелем
При проведении исследований веществ с предполагаемым составом, применяют метод хроматографирования со свидетелем - известным веществом. Этот метод используется кода трудно выдержать условия хроматографирования, нет литературных данных Rf для данной системы или адсорбента, использование градиентного метода и т.д. Да и при проведении цветных реакций можно сравнить не только цвета, но и оттенки исследуемых веществ и свидетелей, что также немаловажно. [3]
С другой стороны этот метод требует дополнительных расходов на свидетели.
1.4.15 Физико-химические методы идентификации
Прелесть тонкослойной хроматографии состоит в том, что после хроматографирования каждое разделенное вещество можно в дальнейшем исследовать другими методами гораздо проще. И дело тут не в том, что другие методы хроматографирования не могут этого. Дело тут в сложности выделения и материальных затратах на специальные приспособления, у которых только одна задача - выделить вещество.
В тонкослойной хроматографии есть только одна трудность - снять слой сорбента и вымыть из него вещество. В дальнейшем можно его исследовать с использованием ИК и УФ-спектрометрии, рентгено-структурными методами, ЯМР и т.д.
Поэтому, используя тонкослойную хроматографию для разделения смесей, можно не только исследовать каждый компонент различными методами, но и наработать небольшое количество, в том числе и для свидетелей. [4]
1.4.16 Методы количественного анализа
Количественный анализ в тонкослойной хроматографии имеет несколько видов, характеризующий каждый этап развития метода. И хотя некоторые методы можно применять только как полуколичественные, они до сих пор применяются на практике.
Метод визуального сравнения. Как говорилось выше, интенсивность окраски пятна и его размер от количества хроматографируемого вещества. Поэтому визуальное количественное определение построено на нескольких приемах.
Метод разбавления. Этот метод заключается в том, что для каждого вещества определяют предельную концентрацию, при которой вещество не может быть определено хроматографическим методом. При хроматографировании исследуемого вещества поводят разбавление до тех пор, пока оно перестает проявляться на пластинке. Содержание вещества С, определенное таким методом находят по формуле:
C = an
где n – разбавление, а – концентрация вещества, при котором оно не проявляется при хроматографировании.
Метод определения площади пятна. Если наносить одинаковые объемы исследуемых веществ и свидетелей, то получившиеся после хроматографирования площади пятен пропорциональна логарифму концентрации вещества.
S= alnc + b
где а и b - эмпирические коэффициенты, определяемые экспериментальным путем.
Если пятно разделенного вещества имеет резкие границы, то площадь пятна можно определить весовым методом (вырезать пятно и взвесить), замеряется планиметром. Этот метод дает ошибку до 10-15%.
Однако он имеет ряд существенных недостатков. Первый и самый существенный в том, что таким образом можно определять концентрацию окрашенных веществ или имеющих флуоресценцию в УФ области (254, 366 нм). Этот недостаток можно устранить добавлением в сорбент различных люминофоров, то при этом увеличивается погрешность определения. Обработка пластин проявляющими веществами (реактивами) также может быть использована (например использование фильтровальной бумаги пропитанной проявляющим реагентом с последующим контактом с хроматографической пластинкой и дальнейшим определением на ней площади проявленного вещества), но погрешность определения также высока.
Необходимость более достоверного результата количественного определения привела к использованию инструментальных методов.
Метод элюирования. Этот метод заключается в том, что разделенное вещество смывают с сорбента растворителем и определяют его концентрацию уже другими методами - фотометрическими, полярографическими и т.д. Это достаточно точный метод, но только при условии количественного выделения разделенного вещества. Из-за высокой трудоемкости метод используется достаточно редко и неприемлем при большом количестве исследуемых образцов.
Фотографический метод определения заключается в фотографировании пластинок с разделенным веществом и дальнейшим определением степени почернения, с использованием десинтометров.
Радиографический метод аналогичен фотометрическому, только с той разницей, что определяется почернение пластинки, вызванное излучением разделенного вещества. Этот метод используется только при определении веществ с мечеными атомами.
Фотодесинтометрический метод может быть использован без выделения вещества с пластинки и основан на определении не только площади пятна, но и его интенсивности.
Это наиболее точный метод определения концентрации веществ, так как позволяет при использовании калибровочных графиков, проводить достаточно точные количественные определения всех разделенных веществ (до 2-10%) непосредственно на пластинке за короткий промежуток времени.
Неудивительно, что при развитии тонкослойной хроматографии, применение десинтометров увеличивается, чувствительность и, следовательно, точность определения концентрации разделенных веществ повышается и приближается к точности высокоэффективной жидкостной хроматографии. [Алесковский В.Б., Бардин В.В., Булатов М.И. Физико-химические методы анализа. Практическое руководство.- Л.: Химия, 1988. - 376с.]
1.4.17 Способы проведения тсх
Подготовка пробы является очень ответственной операцией в ТСХ. Размывание в ТСХ в первую очередь связано с качеством нанесения стартовой зоны образца.
Выбор растворителя и способы нанесения пробы
К растворителю, из которого пробу наносят на слой сорбента, предъявляют следующие требования: полная растворимость в нем всех компонентов пробы; относительная летучесть (для быстрого удаления с пластины перед началом разделения); низкое значение Rf (при использовании полярных растворителей, в которых Rf разделяемых веществ близко к 1, пробы хроматографируются непосредственно при нанесении, что искажает форму стартового пятна и может влиять на Rf, особенно при ТСХ в менее полярных элюентах); хорошая смачиваемость слоя (важно для ОФТСХ).
Стартовая зона должна быть по возможности минимальной, в особенности для ВЭТСХ. Слишком высокая концентрация вещества в стартовом пятне может замедлить растворение пробы при элюировании.
Для нанесения проб используют стеклянные платино-иридиевые капилляры, микропипетки, шприцы, а также специальные дозирующие устройства, например, платино-иридиевые капилляры с максимальным объемом дозирования 22 нл на 1 м длины. При отборе одной и той же пробы капилляром воспроизводимость введения пробы составляет ±0,7% от ее объема.
При нанесении вязких проб можно нагревать пластины или подсушивать их феном для непрерывного испарения растворителя. Использование двухфазных пластин с предадсорбционным слоем снимает многие проблемы. На границе двух сорбентов зона сжимается в виде узкой полосы независимо от качества нанесения стартового пятна (рис. 2).
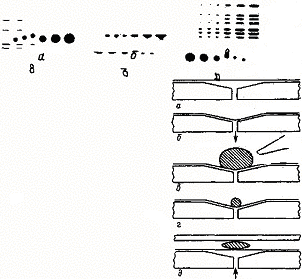
Рис. 2. ТСХ смеси красителей на двухфазных пластинах с предадсорбционным слоем.
Рис. 3. Последовательность операций при нанесении образца контактным способом
Пробу можно сконцентрировать в узкую стартовую зону при погружении пластины в растворитель, для которого значения Rf всех компонентов близки к 1, и пропусканием его несколько выше зоны нанесения пробы, после чего элюирование прекращают, а пластину быстро высушивают. Подобную операцию повторяют несколько раз. Для вязких и разбавленных проб эффективен способ контактного нанесения. На рис. 3 показана последовательность операций нанесения пробы по этому методу:
а) над отверстием в металлической пластине помещают смоченную перфторкосином этиленпропиленовую пленку, которая не смачивается никакими растворителями;
б) с помощью вакуумирования пленка плотно прижимается к пластине и слегка втягивается в отверстие, образуя углубление;
в) в углубление вносят пробу, которая остается там в виде шарообразной капли, так как не смачивает пленку;
г) растворитель испаряют, подсушивая сверху феном, пока шарик не уменьшится до нужного размера;
д) пластину помещают слоем вниз на пленку над пробой и, заменяя вакуум легким давлением, переносят на нее пробу.
Этот метод позволяет нанести на пластину одновременно несколько образцов без их предварительного растворения и концентрировать разбавленные пробы.
Существенным является постоянство расстояния линии нанесения проб от нижнего края пластины и линии погружения пластины в элюент.
Линейная, круговая и антикруговая ТСХ. В колоночной жидкостной хроматографии возможно только линейное движение элюента. В ТСХ благодаря открытому слою сорбента можно осуществить три типа элюирования: линейное, круговое и антикруговое (рис. 4). Кроме того, в ТСХ можно реализовать дополнительно различные методики, позволяющие значительно увеличивать пиковую емкость.
Наиболее широко используют линейное развитие хроматограмм. Пробы наносят на стартовую линию параллельно одной из сторон пластины. Пластину помещают вертикально в хромато-графическую камеру, на дно которой налит элюент, и проводят восходящую ТСХ. В линейной ТСХ с увеличением Rf, вещества размывание увеличивается. Линейное развитие хроматограмм можно осуществить и при горизонтальном положении пластины
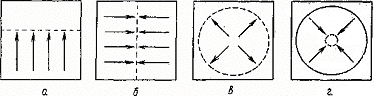
Рис. 4. Типы элюирования в ТСХ: а – линейное; б – линейное (горизонтальное); в – круговое; г – антикруговое с подачей на нее элюента с одной или с обеих сторон.
Для восходящей линейной ТСХ в качестве хроматографической камеры можно использовать любой сосуд прямоугольной или цилиндрической формы. Однако в таких камерах трудно получить воспроизводимые результаты, так как по мере поднятия элюента по пластине происходят фронтальное разделение элюента в слое сорбента (полизональная ТСХ), адсорбция и капиллярная конденсация компонентов элюента из паровой фазы на сухую часть пластины и другие процессы. В подобных камерах, можно проводить ТСХ с насыщением (стенки камеры покрывают фильтровальной бумагой, смоченной элюентом) или без насыщения.
Для уменьшения влияния паровой фазы в линейной ТСХ используют «сэндвич»-пластины. С трех сторон двух пластин снимают узкий слой сорбента и, проложив между ними прокладку (толщина 0,5—2 мм), плотно скрепляют такой «сэндвич»-зажимом и опускают в камеру с элюентом. ТСХ проводят, таким образом, одновременно на двух пластинах, расположенных слоем друг к другу. В качестве второй пластины можно использовать, стеклянную подложку без слоя адсорбента.
В круговой ТСХ пробы наносят на некотором расстоянии от центра пластины по окружности, а элюент подают в центр (рис. 4, б). Оптимальное разрешение достигается круговой хроматографией при К' = 100 (Rf = 0,009), т. е. хорошо разделяются вещества с низкими значениями Rf.
В антикруговой ТСХ пробы наносят по окружности по периферии пластины и элюент подают в направлении к центру пластины (рис. 4, г). При этом хроматографические зоны сжимаются по мере движения по пластине. В антикруговой ТСХ одновременно можно анализировать наибольшее количество проб. Это наиболее быстрый метод ТСХ. Антикруговая ТСХ дает хорошие результаты при разделении веществ с высокими значениями Rf. Для реализации круговой и антикруговой ТСХ фирма «Камаг» выпускает специальные так называемые U-камеры. Применение U-камер позволяет повысить воспроизводимость результатов ТСХ за счет элиминирования влияния паровой фазы и возможности проведения ТСХ в атмосфере инертного газа. Кроме того, имеется возможность наносить пробу в поток элюента. Круговую и антикруговую ТСХ можно реализовать и более простым способом при использовании, например, чашки Петри для круговой ТСХ и 2 чашек Петри, вставленных одна в другую для антикруговой ТСХ. Для увеличения пиковой емкости в ТСХ используют методы проточной, многократной, градиентной и двумерной ТСХ. [1]
Глава 2. Контроль качества пищевых продуктов посредством метода ТСХ
2.1 Определение ддт, ддэ, ддд, альдрина, дильдрина, гептахлора, кельтана, метоксихлора, эфирсульфоната и других ядохимикатов в продуктах питания хроматографией в тонком слое
Принцип метода. Метод основан на извлечении препаратов из пробы органическими растворителями (н-гексан, петролейный эфир, диэтиловый эфир, хлороформ), очистке экстракта и последующем хроматографировании в тонком слое окиси алюминия или силикагеля. Подвижным растворителем служит нормальный гексан или гексан в смеси с ацетоном. Места локализации препаратов обнаруживают после опрыскивания пластинок раствором аммиаката серебра в ацетоне или 2%-ным раствором дифениламина с последующим ультрафиолетовым облучением. Количественное определение проводится путем визуального сравнения или измерения площадей пятен проб и стандартных растворов. [5]
Минимально детектируемое количество при проявлении азотнокислым серебром 0,5 мкг, при проявлении дифениламином 10 мкг. Чувствительность определения в воде и вине 0,002 мг/л, яблоках и капусте 0,02, в других овощах и фруктах 0,01, зерне 0,05, сене 0,05, рыбе, мясе, внутренних органах, жире 0,02, молоке, твороге 0,01, почве 0,01 мг/кг. Полнота определения 90±5%.
Реактивы и растворы: Гексан «х. ч.», петролейный эфир (tкип. 40—70еС), диэтиловый эфир для наркоза, хлороформ х. ч., бензол х. ч., ацетон х. ч., натрий сернокислый безводный х. ч., насыщенный раствор безводного сернокислого натрия в серной кислоте плотностью 1,84 г/см3 (100 г безводного сернокислого натрия растворяют в 1л серной кислоты), окись алюминия для хроматографии или силикагель КСК, просеянные через сито 100 меш., кальций сернокислый (CaSO4×2H2O) «ч. д. а.» - просушивают в сушильном шкафу 6 ч при 150—160° С и хранят а банке с притертой пробкой; спирт этиловый.
Проявляющий реактив №1. 0,5 г азотнокислого серебра растворяют в 5 мл дистиллированной воды, прибавляют 7 мл аммиака плотностью 0,9 г/см3 и доводят объем раствора до 100 мл ацетоном. Готовят в день употребления. На пластинку 9 ´ 12 см расходуется 8—10 мл раствора.
Проявляющий реактив №2. 2%-й раствор дифениламина в ацетоне. 5%-ный водный раствор оксалата калия ч. д. а.Насыщенный водный раствор натрия хлористого. Силикагель АСК Воскресенского химкомбината (Московская обл.). Стандартные растворы пестицидов (100—200 мкг/мл). 10—20 мг ДДТ х. ч. или др. пестицида растворяют в 100 мл гексана, хранят в холодильнике в плотно закрытой склянке. Пластинки для хроматографии. Тщательно промытую содой, хромовой смесью, дистиллированной водой, высушенную стеклянную пластинку 9 ´ 12 или 13 ´ 18 см протирают этиловым спиртом или эфиром и покрывают сорбционной массой. Массу готовят следующим образом: 50 г просеянной через сито 100 меш окиси алюминия смешивают в фарфоровой ступке с 5 г сернокислого кальция, прибавляют 75 мл дистиллированной воды и перемешивают в ступке или колбе до образования однородной массы. 10 г сорбционной массы равномерно распределяют по всей поверхности пластинки 9 ´ 12 см путем ее покачивания. Сушат пластинки при комнатной температуре 18—20 ч и хранят в эксикаторе.
Сорбционную массу можно приготовить из силикагеля (35 г силикагеля, 2 г гипса и 90 мл дистиллированной воды). Силикагель предварительно очищают от примесей, - заливают на 18—20 ч. разбавленной соляной кислотой (1 : 1), кислоту сливают, промывают силикагель водой и кипятят 2—3 ч с разбавленной азотной кислотой (1 : 1), промывают проточной водопроводной, затем дистиллированной содой до нейтральной реакции промывных вод, сушат в сушильном шкафу 4—6 ч при температуре 130° С. Силикагель дробят и просеивают через сито 100 меш, хранят в склянке с притертой пробкой.
Делительные воронки на 100, 250 и 500 мл.
Мерные колбы на 100 мл.
Колбы с притертыми пробками на 250 и 500 мл.
Прибор для отгонки растворителей.
Колбы круглодонные на 50 и 100 мл.
Пульверизаторы стеклянные для опрыскивания пластинок (рис. 1).
Камера для хроматографирования. Стеклянный сосуд с притертой крышкой. Можно использовать эксикатор.
Камера для опрыскивания.
Микропипетки для нанесения стандартных растворов.
Пипетки или шприцы для нанесения проб.
Бани водяные.
Ртутно-кварцевая лампа.
Хроматографические колонки 18 ´ 390 мм.
Ход анализа
Пищевые продукты и корма. Для анализа из средней пробы отбирают: овощи, фрукты, грибы, зерно, творог — 500 г, сыр— 100, мясо, рыба — 250, масло сливочное, сливки — 200, трава — 200, сухие грибы, сено — 50 г, вино, вода, молоко — 500 мл. Масло растапливают, овощи, фрукты и т.п. измельчают и тщательно перемешивают.
Вода, вино. Пробы отбирают только в стеклянную посуду. 200—300 мл пробы в колбе с притертой пробкой экстрагируют в течение 15 мин на приборе для встряхивания н-гексаном или петролейным эфиром тремя порциями по 30 мл или диэтиловым эфиром тремя порциями по 40—50 мл. Экстракцию можно проводить в делительных воронках, энергично встряхивая жидкость 3 раза по 3—5 мин.
В объединенные экстракты насыпают около 10 г безводного сернокислого натрия или объединенные экстракты фильтруют через воронку с сернокислым натрием. Экстракты переносят в прибор для отгонки растворителей, отгоняют растворитель до небольшого объема (0,1—0,2 мл).
Овощи, фрукты. 10г измельченной пробы трижды экстрагируют в течение 15 мин на аппарате для встряхивания н-гексаном или петролейным эфиром тремя порциями по 30 мл. Объединенные экстракты сушат безводным сернокислым натрием, переносят в прибор для отгонки растворителей, отгоняют растворитель до небольшого объема.
Зерно, грибы. 10 г зерна, 50 г сырых или 10 г сухих грибов мелко измельчают и экстрагируют па приборе для встряхивания н-гексаном или петролейным эфиром порциями по 30 мл. Объединенные экстракты переносят в делительную воронку, прибавляют 10 мл насыщенного раствора безводного сернокислого натрия в серной кислоте и осторожно встряхивают несколько раз. Отделяют органический слой и повторяют обработку до тех пор, пока кислота не станет бесцветной. Сушат экстракт над безводным сернокислым натрием, отгоняют растворитель до небольшого объема.
Яблоки, капуста, трава, сено. 10 г пробы яблок, 10— 20 г капусты, 10—20 г травы или 5—10 г сена измельчают, заливают 100 мл ацетона. Встряхивают 2—3 мин, прибавляют 20 мл ледяной дистиллированной воды и охлаждают на льду 30 мин. Экстракт сливают и фильтруют холодным, экстракцию повторяют. Из объединенных водно-ацетоновых экстрактов отгоняют ацетон, а из водного слоя экстрагируют препарат н-гексаном или петролейным эфиром тремя порциями по 10 мл в течение 10 мин. Гексановые экстракты очищают серной кислотой, насыщенной безводным сернокислым натрием. Сушат безводным сернокислым натрием. Отгоняют растворитель до небольшого объема и наносят на пластинку. Если очистка неполная, экстракт испаряют досуха, остаток промывают холодным ацетоном 3 раза порциями по 0,2 мл и сразу наносят на пластинку.
Рыба, мясо. Мясо пропускают через мясорубку. Рыбу очищают от чешуи, внутренних органов и пропускают через мясорубку. 25 г пробы заливают 40 мл н-гексана или петролейного эфира, взбалтывают 2 мин, разбивают образовавшиеся комочки и оставляют на 30 мин. Экстракцию повторяют дважды. Объединенные экстракты переносят в делительную воронку, добавляют 10 мл насыщенного раствора сернокислого натрия в серной кислоте и осторожно встряхивают несколько раз. Отделяют органический слой. Обработку повторяют до тех пор, пока кислота не станет бесцветной. Сушат экстракты безводным сернокислым натрием, растворитель испаряют до 0,1 мл.
Внутренние органы, животный жир. 25 г измельченной пробы помещают в колбу и заливают концентрированной серной кислотой, насыщенной безводным сернокислым натрием (проба должна полностью находиться в кислоте). Содержимое колбы перемешивают стеклянной палочкой, прибавляют 40 мл н-гексана, непрерывно встряхивают 2 мин, периодически встряхивают в течение 30 мин. Экстракт декантируют, экстракцию повторяют. Слитые в делительную воронку экстракты осторожно встряхивают с концентрированной серной кислотой, насыщенной сернокислым натрием. Операцию повторяют несколько раз до получения бесцветного раствора серной кислоты. Отделяют гексановый слой, сушат безводным сернокислым натрием и испаряют.
Растительные масла. 10 мл масла наливают в делительную воронку, прибавляют 20 мл ацетона, встряхивают и приливают 10 мл этилового спирта. Выделившееся масло сливают. К ацетоно-спиртовому экстракту прибавляют 100 мл 2%-ного водного раствора сернокислого натрия. Из смеси экстрагируют препарат 2 раза н-гексаном порциями по 30 мл, каждый раз встряхивая 5 мин. К объединенным гексановым экстрактам прибавляют 20 мл насыщенного раствора безводного сернокислого натрия в серной кислоте и осторожно встряхивают несколько раз. Отделяют органический слой и повторяют обработку до тех пор, пока кислота не обесцветится. Сушат экстракт и испаряют растворитель до 0,1—0,2 мл.
Молоко, сливки. К 50 мл молока или 20г сливок прибавляют концентрированную серную кислоту (30—40 мл) до полного почернения пробы. Охлажденный до 10—15° С раствор переносят в делительную воронку и экстрагируют н-гексаном 2 раза порциями по 25 мл. Для полного извлечения воронку встряхивают 2 мин, затем оставляют ее на несколько минут до полного разделения слоев. Если образуется эмульсия, прибавляют 1—2 мл этилового спирта. К объединенным экстрактам в делительной воронке прибавляют 10 мл концентрированной серной кислоты, насыщенной сернокислым натрием, и осторожно встряхивают несколько раз. Очистку повторяют до получения бесцветной кислоты.
Творог, сыр. 50 г творога или 10 г измельченного на терке сыра заливают 40 мл н-гексана или петролейного эфира, непрерывно встряхивают 2—3 мин и периодически встряхивают в течение 30 мин. Экстракцию повторяют. Объединенные экстракты в делительной воронке очищают серной кислотой, как указано выше.
Сливочное масло. 20 г сливочного масла растапливают на водяной бане в круглодонной колбе, прибавляют 50 мл ацетона, тщательно перемешивают до растворения жира, прибавляют 10 мл ледяной дистиллированной воды и охлаждают па льду до затвердения жира (примерно 30 мин). Сливают ацетоновый экстракт процедуру повторяют 3 раза. Из объединенных экстрактов в круглодонной колбе ацетон отгоняют на водяной бане. Пестициды экстрагируют из оставшегося водного экстракта гексаном тремя порциями по 10 мл в течение 5 мин. Объединенные гексановые экстракты в делительной воронке обрабатывают серной кислотой с сернокислым натрием. Очищенный экстракт сушат и упаривают. Второй способ подготовки проб молока и молочных продуктов. Молоко (кефир, простокваша, кумыс и другие цельномолочные продукты). 25 мл продукта помещают в делительную воронку на 300 мл, приливают по 5 мл раствора оксалата калия и насыщенного раствора NaCl, перемешивают, приливают 100 мл ацетона, встряхивают 2 мин. Приливают 100 мл хлороформа и встряхивают 2 мин. Воронку оставляют до полного разделения слоев. Верхнюю фазу отбрасывают, а нижнюю выливают в круглодонную колбу со шлифом и испаряют растворитель досуха. Остаток смывают 30 мл гексана.
Сгущенное молоко. 10—20%-ные сливки. К 10 г продукта прибавляют 10 мл насыщенного раствора NaCI и выливают в делительную воронку вместимостью 150 мл. К смеси приливают 40 мл ацетона, встряхивают 2 мин, приливают 60 мл хлороформа, встряхивают 2—3 мин и оставляют до разделения фаз. Далее поступают, как при определении пестицидов в молоке.
Сгущенные молочные продукты. 10 г продукта помещают в стаканчик, заливают 10 мл воды с температурой 45—50° С, перемешивают и переносят в делительную воронку емкостью 150 мл, добавляют 5 мл раствора оксалата калия. Содержимое воронки перемешивают, приливают 80 мл ацетона и встряхивают 2—3 мин, добавляют 100 мл хлороформа и встряхивают 5—7 мин. После разделения фаз нижнюю фазу сливают в круглодонную колбу, растворители отгоняют, а остаток растворяют в 30 мл петролейного эфира.
Сухие молочные продукты. 3 г сухих молочных продуктов (2 г сухих сливок) высыпают в стаканчик, приливают 15 мл дистиллированной воды с температурой 40—45° С, размешивают и переносят в делительную воронку вместимостью 300 мл, приливают по 5 мл раствора оксалата калия и насыщенного раствора поваренной соли. Содержимое воронки перемешивают, добавляют 80 мл ацетона и встряхивают 3—5 мин, приливают 100 мл хлороформа, встряхивают 5 мин и оставляют на 3—5 мин (до разделения фаз). Нижнюю фазу сливают в колбу, растворители отгоняют, а остаток смывают 30 мл н-гексана.
Масло, молочный жир. 2 г продукта высыпают в стаканчик, подогревают до 40° С и растворяют в 30 мл н-гексана.
Сметана, 30—40%-ные сливки. 5 г продукта отвешивают в стаканчик, добавляют 10 мл насыщенного раствора поваренной соли и переносят в делительную воронку вместимостью 150 мл. Стаканчик обмывают 40 мл ацетона, смывы переносят в делительную воронку, которую встряхивают 1—2 мин, добавляют 70 мл хлороформа и встряхивают 2—3 мин. Воронку оставляют на 3—5 мин (до разделения фаз), нижнюю фазу слипают в колбу для отгонки растворителей, растворители отгоняют, а остаток смывают 30 мл л-гексана.
Творог, сыр. 10 г творога или измельченного на терке сыра растирают с 10 мл насыщенного раствора поваренной соли и переносят в делительную воронку на 250—300 мл. Прибавляют 80 мл ацетона, встряхивают 2 мин, прибавляют 100 мл хлороформа и вновь встряхивают. Нижнюю фазу используют для анализа после отгонки растворителей, растворив остаток в 30 мл н-гексана.
Очистка экстракта от молочного жира. В нижнюю часть хроматографической колонки помещают стекловату, затем насыпают в колонку 70 мл силикагеля АСК и уплотняют его постукиванием. Колонку промывают 50 мл гексана, а прошедший через нее растворитель отбрасывают. На подготовленную таким образом колонку наносят 30 мл полученного из молока или молочных продуктов экстракта. После впитывания экстракта в сорбент пестицид элюируют 110мл смеси бензола с гексаном (3:8) порциями по 25—30 мл. Элюат собирают в круглодонную колбу со шлифом вместимостью 250—300 мл. Через 10 мин после впитывания последней порции растворителя сорбент отжимают с помощью резиновой груши. После очистки растворители отгоняют под вакуумом.
Хроматографирование. На хроматографическую пластинку на расстоянии 1,5 см от ее края шприцем или пипеткой наносят исследуемую пробу в одну точку так, чтобы диаметр пятна не превышал 1 см.
Остаток экстракта в колбочке смывают тремя небольшими порциями (по 0,2 мл) диэтилового эфира, которые наносят в центр первого пятна. Справа и слева от пробы на расстоянии 2 см наносят стандартные растворы, содержащие 1,5 и 10 мкг исследуемых препаратов. Для нанесения 1 мкг стандартный раствор следует разбавлять в 10 раз.
Пластинку с нанесенными растворами помещают в камеру для хроматографирования, на дно которой за 30 мин до начала хроматографирования наливают подвижный растворитель н-гексан, а для препаратов, у которых Rf в гексане ниже 0,3 — смесь гексана с ацетоном (6:1). Край пластинки с нанесенными растворами может быть погружен в подвижный растворитель не более чем на 0,5 см.
После того как фронт растворителя поднимется на 10 см, пластинку вынимают из камеры и оставляют на несколько минут для испарения растворителя. Далее пластинку орошают проявляющим реактивом и подвергают УФ-облучению 10—15 мин (лампа ПРК-4). Пластинки следует располагать на расстоянии 20 см от источника света.
При наличии хлорорганических пестицидов на пластинке появляются пятна серо-черного цвета. Количественное определение осуществляют сравнением размеров пятен пробы и стандартного раствора (визуально или измеряют площади). Между количеством препарата в пробе, не превышающем 20 мкг, и площадью его пятна на пластинке существует прямая пропорциональность. При большем содержании препарата следует использовать пропорциональную часть исследуемого экстракта.
Расчет количества препарата в пробе вычисляют по формуле:
X = A / P или

где X — содержание препарата в пробе, мг/кг или мг/л; А — количество препарата, найденное путем визуального сравнения со стандартным раствором, мкг; Р — масса или объем исследуемой пробы, г или мл; А1 – содержание препарата в стандартном растворе, мкг; S1 – площадь пятна стандартного раствора, мм2; S – площадь пятна пробы, мм2.
Величина Rf зависит от адсорбента, размера его частиц, количества и природы связующего материала, толщины слоя, природы подвижного растворителя, степени насыщения камеры для хроматографирования парами подвижного растворителя, расстояния пройденного растворителем, температуры, количества и природы коэкстрактивных веществ.
Для ДДТ Rf для оксида алюминия составляет 0,61-0,71 (в зависимости от изомера) и 0,50-0,54 на силикагеле.
На слое оксида алюминия обнаруживаются три пятна, которые соответствуют 4,4'-ДДТ (основное пятно), 2,4'-ДДТ и 4,4'-ДДД (слабое пятно). Пятно ДДД проявляется, если содержание ДДТ в анализируемой пробе превышает 20 мкг. Содержание изомеров ДДТ суммируют. На хроматограммах проб пищевых продуктов, содержащих остаточные количества ДДТ, кроме упомянутых трех пятен, обнаруживается четвертое пятно, соответствующее продукту разложения ДДТ — ДДЭ.
На слое силикагеля технический препарат ДДТ дает одно пятно, соответствующее сумме изомеров. Если количество ДДТ в пробе превышает 20 мкг, на хроматограмме появляется слабое пятно, соответствующее ДДД. [5]
2.2 Определение о,о-диэтил-8-(6-хлорбензоксазолинил-3-метил)-дитиофосфата (фозалона) в яблоках методом тонкослойной хроматографии
Фозалон применяется для борьбы с вредителями сада. [6]
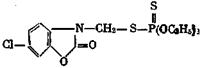
В литературе описана идентификация фозалона и определение его в растениях колориметрическим и биологическим методами [7].
Однако эти методы весьма громоздки, так как требуют сложной очистки растительных экстрактов. Нами был разработан простой качественный и количественный метод определения остаточных количеств фозалона в яблоках с применением тонкослойной хроматографии. Исходным веществом служил чистый препарат фозалона. Опыты проводили с яблоками сорта Ренет Симиренко.
После испытания ряда экстрагентов для извлечения фозалона из яблок был избран хлороформ. Для идентификации фозалона применили метод тонкослойной хроматографии.
При подборе подвижной фазы был изучен ряд органических растворителей и их смесей. Наилучшей для идентификации фозалона является смесь гексана с ацетоном в соотношении 4:1.
Для проявления фозалона на хроматографических пластинках был испытан ряд реактивов, рекомендованных для проявления фосфорорганических пестицидов [8 – 11].
Наилучшие результаты дали 0,2%-ный раствор хлористого палладия [L. Fishbein, J. Fowkes, P. Jones. J. Chromatogr., 23, 47b (1966).] или смесь 0,05 %-ного раствора бром-фенолового синего в ацетоне с 1%-ным водным раствором AgNО3 в соотношении 1:1, с последующим отбеливанием хроматограмм 2,5%-ным раствором лимонной кислоты [Л. М. Фукельман. Сборник трудов ВИЗР, вып. 20, ч. 4, 61 (1964).]. Эти проявители позволяют обнаруживать до 1 мкг фозалона.

Рис. 5. Тонкослойная хроматограмма фозалона1 из экстракта яблок на силикагеле КСК. а – место нанесения экстракта; б – точка нанесения стандарта; в – место отложения фозалона
Рис. 6. Двухмерная тонкослойная хроматограмма фозалона из экстракта яблок на силикагеле КСК. а — б — место нанесения экстракта с содержанием фозалона 50 мкг; в, г, д – точки нанесения стандартных растворов фозалона, соответственно 25, 50 и 75 мкг.
Предлагаемая методика определения фозалона в яблоках заключается в следующем. 100 г яблок измельчают, наносят пипеткой определенное количество фозалона. Экстрагируют хлороформом при встряхивании на шутель-аппарате 1,5 часа. Хлороформный экстракт отделяют от растительной массы фильтрованием. Растительную массу на фильтре промывают трижды хлороформом (по 20 мл). Фильтрат и промывные порции хлороформа собирают в колбу для отгонки растворителя. Хлороформ отгоняют под вакуумом до объема 5—10 мл. Переносят его количественно в стаканчик, упаривают до 0,5 мл и наносят на пластинку полоской шириной 1 см длиной 5 см на расстоянии 1,5 см от нижнего края пластинки.
Для хроматографирования используют стеклянные пластинки размером 9 X 15 см, на которые наносят тонкий слой силикагеля марки КСК (40 меш), закрепленный гипсом.
Хроматографирование проводят в системе гексан — ацетон (4:1).
Пластинки проявляют указанными выше проявителями. Причем в случае применения хлористого палладия пятна фозалона окрашиваются в желтый цвет на белом фоне, а примеси —.в коричневый. Окраска не исчезает в течение длительного промежутка времени. При применении второго проявителя пятна фозалона окрашиваются в синий цвет на желтом фоне, а примеси совсем не окрашиваются. Следует отметить, что в этом случае пятна фозалона через 10—15 мин. бледнеют. Rf фозалона равно 0,5 (рис. 5).
Чувствительность метода 0,1 мг/кг.
Этот вариант метода позволяет качественно определять содержание фозалона в растительной пробе.
Для количественного определения фозалона в пробе мы использовали двухмерную хроматографию на пластинках размером 15 ´ 20 см.
Экстракцию и отгонку проводят так же, как и в предыдущем случае. Экстракт без очистки наносят на пластинку полоской длиной 5 см и шириной 1 см на расстоянии 1,5 см от нижнего края (нижняя сторона пластинки 15 см) и 2 см от левого края пластинки.
Хроматографирование проводят в подвижном растворителе гексан — ацетон (4 : 1). Дают растворителю продвинуться на 12—15 см, потом пластинку высушивают на воздухе в течение 20 мин. Далее поворачивают ее на 90е так, чтобы левый край пластинки служил основанием. С левой стороны пластинки наносят стандарты с разным содержанием фозалона и хроматографируют в том же подвижном растворителе. С целью получения хорошо сформированных пятен проводят повторное хроматографирование в том же направлении, но с применением другого подвижного растворителя — хлороформа.
После проявления пластинок теми же реактивами, что и в предыдущем случае, получали пятна с Rf = 0,82 (рис. 6). Количественное определение фозалона проводят сравнением пятна анализируемой пробы с пятнами стандартов.
Предложенным методом было проведено определение остатков фозалона в яблоках (см. таблицу).
Определение фозалона в 100 г яблок
| Внесено фозалона, мкг | Найдено фозалона, мкг | Ошибка | Внесено фозалона, мкг | Найдено фозалона, мкг | Ошибка | ||
| мкг | % | мкг | % | ||||
| 25 | 20 | 5 | - 20 | 75 | 70 | 5 | - 6,7 |
| 50 | 45 | 5 | -10 | 100 | 85 | 15 | - 15 |
| 50 | 40 | 10 | - 20 | ||||
Глава 3. ТСХ – оборудование
3.1 Оборудование фирмы НТЦ ЛЕНХРОМ для инструментальной ТСХ
Оборудование фирмы НТЦ ЛЕНХРОМ позволяет провести все рабочие этапы современной тонкослойной хроматографии:
- дозировка и нанесение проб
- проведение процесса хроматографии
- УФ-обнаружение
- детектирование (окрашивание) хроматограмм
- видеозапись изображений пластинок
- количественная обработка хроматограмм
Объекты исследования и области применения планарной хроматографии
1. Готовые лекарственные формы
- подлинность действующих веществ
- точность дозировки
- наличие вредных примесей и добавок
2. Фармакология
- фармакокинетические исследования
- изучение метаболизма действующих веществ
3. Биохимия и медицина
- определение содержания важнейших биологически активных соединений в жидкостях и тканях организма
- изменение содержания этих веществ при патологии
- судмедэкспертиза
- допинг-контроль
4. Охрана окружающей среды
- анализ пестицидов в питьевой и сточных водах, осадках, продуктах питания
- анализ токсических веществ (например, афлатоксинов) в продуктах питания
3.1.1 Портативный набор для тсх
![]() Портативный набор для ТСХКомплект предназначен для
качественного и полуколичественного анализов многокомпонентных смесей органических
и неорганических веществ и может найти применение во всех областях науки и
техники, где для анализа используется метод тонкослойной хроматографии.Комплект позволяет:
Портативный набор для ТСХКомплект предназначен для
качественного и полуколичественного анализов многокомпонентных смесей органических
и неорганических веществ и может найти применение во всех областях науки и
техники, где для анализа используется метод тонкослойной хроматографии.Комплект позволяет:
- проводить анализ смесей методом ТСХ, основанном на разделении анализируемых веществ за счет различной скорости перемещения их по тонкому слою сорбента, нанесенного на плоскую подложку (стеклянную, полимерную или алюминиевую);
- наносить точное количество анализируемых веществ на хроматографические пластины;
- анализировать одновременно большое количество проб;
- обнаруживать и просматривать хроматограммы в ультрафиолетовом свете;
- окрашивать хроматограммы;
- удалять органические растворители с хроматограмм высушивающим устройством с подогревом.
Комплект включает:
- Ультрафиолетовый осветитель УФО-254
- Хроматографическую камеру для пластин размером 130-130-56 см
- Пластины для ТСХ (10х10), одна упаковка - 50 шт
- Пульверизатор для вязких и агрессивных жидкостей
- Пульверизатор для маловязких и неагрессивных жидкостей
- Фен
Торрированные стеклянные капилляры V=1 ml
3.1.2 Базовый набор для ТСХ
Базовый набор для ТСХ (расширенный)
- УФ-кабинет 254/365 (ультрафиолетовая лампа) - 1 шт.
- Спрей-камера коррозионностойкая (камера для окрашивания хроматограмм методом опрыскивания) - 1 шт.
- Столик с подогревом для нанесения образцов на пластины - 1 шт.
- Камера хроматографическая (для пластин 15x15 см) - 2 шт.
- Камера хроматографическая (для пластин 10x10 см) - 2 шт.
- Сушильный шкаф ШСУ - 1 шт.
- Пульверизатор для агрессивных жидкостей - 1 шт.
- Пульверизатор для маловязких жидкостей с резиновой "грушей"- 1 шт.
- Микрошприц V=1 мкл - 1 шт.
- Микрошприц V=10 мкл - 1 шт.
- Стеклянные капилляры V=1 мкл -100 шт.
- Микрокап (держатель капилляров) - 1 шт.
- Пластины на стеклянной подложке АТСХ (10x10 см) - 20 шт.
- Пластины "Сорбфил" ПТСХ-П-А 10x10 см - 50 шт.
- Пластины "Сорбфил" ПТСХ-П-А-УФ 10x10 см - 50 шт.
- Пластины "Сорбфил" ПТСХ-АФ-А 10x10 см - 50 шт.
- Пластины "Сорбфил" ПТСХ-АФ-А-УФ 10x10 см - 50 шт.
3.1.3 Оборудование для проведения ВЭТСХ анализов
Качественный анализ: Набор для ВЭТСХ CAMAG, включая УФ-кабинет
Набор для ВЭТСХ дополнен горизонтальной ВЭТСХ камерой и новым аппликатором CAMAG Nanomat 4, диспенсером капилляров с держателем для одноразовых капилляров объемом 0.5б1б2б и 5 мкл. При использовании горизонтальной камеры количество одновременно анализируемых проб удваивается, а расход растворителей значительно сокращается. В отличие от обычных, ВЭТСХ пластины имеют лучшее разрешение. Предназначен для качественного и количественного анализов (при использовании систем денситометрии).
Система для документирования и видеоденситометрии
Новейшая система для качественного и количественного анализа в ТСХ при детектировании в видимой области спектра, включая режим флуоресценции. Специально разработана для массовых анализов растительного сырья на подлинность или для скриннинга образцов на присутствие "ключевых" компонентов. Полное документирование оцифрованных изображений с хранением в базах данных эталонных изображений, например, для сравнения с рабочими анализами.
Количественный анализ: Полуавтоматический аппликатор CAMAG Linomat 4 для нанесения "полос" способом распыления.
Линомат 4 распыляет раствор пробы в форме узкой полосы на поверхность пластины (ТСХ или ВЭТСХ). Такой способ нанесения дает возможность дозирования большого объема образца по сравнению с контактным методом, а также гарантирует максимально возможное разрешение для выбранной хроматографической системы для любого варианта: качественного, количественного или препаративного разделения.
Техника нанесения, используемая в приборе CAMAG Linomat 4, стала синонимом качества в количественной инструментальной хроматографии.
Спектроденситометр CAMAG Scanner 3
Спектроденситометр третьего поколения с программным управлением от ПЭВМ. Уникальные характеристики оптико-механической части гарантируют высокое разрешение (до 25 мкм) и скорость сканирования. Возможность работы в режиме отражения (УФ и флуоресценция) и пропускания (УФ и флуоресценция). Развитое программное обеспечение с готовыми методами количественного анализа, включая статистические расчеты и методы калибровки. Сертифицирован по ISO и ГОСТ.
Программа-менеджер для планарной хроматографии - winCATS
Новейшее программное обеспечение для документирования всех стадий хроматографии от пробоподготовки, нанесения, элюирования, дериватизации до детектирования. С использованием процедур управления приборами EquiLink программа становится операционной оболочкой для управления всеми приборами, сбора данных и обработки результатов в соответствии с GLP/GMP.
Новый информационные технологии превратили ТСХ в инструментальный метод, дополняющий ВЭЖХ в современной лаборатории.
3.1.4 Пластины многократного пользования для высокоэффективной и аналитической ТСХ
В ИВС АН СССР разработана технология производства пластин для тонкослойной хроматографии на стеклянной, полимерной подложке и подложке из алюминиевой фольги. В настоящее время по этой технологии выпускаются пластины для ТСХ на стеклянной подложке размером 20x20 см и полимерной подложке размером 10x10 см в количестве 50000 и 500000 штук в год, соответственно, в двух модификациях: АТСХ (пластины для аналитической ТСХ) и ВЭТСХ (пластины для высокоэффективной ТСХ). В качестве сорбента используется фракционированный силикагель КСКГ (СССР) с диаметром пор 110-130 А и размером фракций 5-17 мкм (АТСХ) и 8-12 мкм (ВЭТСХ). Толщина слоя сорбента 110-130 мкм. Могут выпускаться пластины с толщиной слоя до 200 мкм. Выпускаются также пластины этих модификаций с люминофором с возбуждением 254 нм.
Особенности ТСХ-пластин, выпускаемых по технологии ИВС, связаны с использованием в качестве связующего золя кремневой кислоты, который после нагревания переходит в силикагель. Таким образом, разработанные ТСХ-пластины не имеют связующего и состоят из двух компонентов: слоя силикагеля и подложки. Если применять стеклянную подложку, то такие ТСХ-пластины являются химически прочными. Их химическая устойчивость определяется химической стойкостью силикагеля. В результате ТСХ-пластины ИВС могут многократно обрабатываться агрессивными реагентами, например, горячей хромовой смесью, что снимает ограничения в использовании коррелирующих реагентов для модификации и детектирования пластин и позволяет проводить многократную (до 10 раз) регенерацию пластин с помощью хромовой смеси. Механическая прочность слоя сорбента может регулироваться, обеспечивая, с одной стороны, транспортировку и многократность обработки пластин и, с другой стороны, возможность экстракции слоя адсорбента с разделившимися веществами для их последующей эволюции.
По эффективности АТСХ- и ВЭТСХ-пластины ИВС соответствуют ДС- и HPTLC-пластинам фирмы "Merck" (ФРГ).
Пластины широко испытаны в анализе производных аминокислот, пестицидов, липидов, антибиотиков, а также других классов органических и неорганических соединений и высокополимеров.
|
|
Пластины на полимерной подложке: - ПТСХ-А, 10x10 - ПТСХ-А-УФ, 10x10 - ПТСХ-А, 10x15 - ПТСХ-А-УФ, 10x15 - ПТСХ-В, 10x10 - ПТСХ-В-УФ, 10x10 Стоимость: 0,5-0,9 $ за 1 шт.
|
||||||||||||






3.1.5 Контроль эффективности разделения
Для идентификации тонкослойных хроматограмм требуется ультрафиолетовое освещение двух типов:
- длинноволновое, с длиной волны 365 нм - под которым разделенные вещества на пластинах флюоресцируют яркими пятнами, часто разного цвета, на темном фоне. Чувствительность детектирования в таком свете увеличивается с ростом интенсивности облучения;
- коротковолновое, с длиной волны 254 нм - под которым вещества, адсорбируя свет, становятся видимыми. Эти вещества выглядят как темные пятна на ярком зеленом фоне пластины.
УФ-кабинеты фирмы НТЦ "Ленхром" разработаны специально для лабораторий ТСХ. Однако, они могут использоваться и для решения других задач: проверки подлинности документов, и денежных знаков, в минералогии, археологии, криминалистике и др. Лампы помещены в защитный кожух, не пропускающий видимый свет, что позволяет исследовать хроматограммы в незатененных помещениях, сохраняя при этом высокую чувствительность.
Выпускаются УФ-кабинеты двух типов: УФ-кабинет254/365, УФО-254.
3.1.6 Хроматографические камеры
Камера с плоским дном и притертой крышкой - классическая емкость для хроматграфии, позволяющая проводить процедуру хроматографии в условиях частичного или полного насыщения атмосферы камеры парами растворителя. Благодаря оптимальным размерам, происходит низкое потребление растворителей в процессе работы. Размер камер предназначен для пластин разных стандартных размеров: 10х10, 15х15 и 20х20 см.
Камера для проявления в парах йода
Камера для окрашивания - плоская стеклянная камера предназначена для окрашивания хроматографических пластинок растворами реагентов в легколетучих растворителях методом погружения. Камера позволяет равномерно по площади окрасить поверхность пластины. Камера потребляет малое количество детектирующего реагента. Камеры предназначены для пластин размером 10х10 и 15х15 см.
3.1.7 Постхроматографическая дериватизация
Пульверизатор для агрессивных жидкостей
Благодаря конструктивным особенностям, пульверизатор безопасен при работе с агрессивными (н-р, концентрированные кислоты и щелочи) жидкостями. Пульверизатор может работать как с использованием резиновой груши (маловязкие жидкости), так и с использованием сжатого воздуха, что позволяет распылять вязкие жидкости.
Пульверизатор для маловязких жидкостейПульверизатор работает с использованием резиновой груши. Пульверизатор используют для распыления маловязких и неагрессивных жидкостей, т.е. водных и спиртовых растворов.
Спрей-камера
Устойчивая к агрессивным средам спрей-камера предназначена для окрашивания хроматограмм методом орошения (опрыскивания) различными детектирующими реагентами. Камера гарантирует безопасность процесса распыления вредных веществ для организма человека. Камера, по желанию, может быть укомплектована гибкой трубой для соединения с вытяжной вентиляцией.
3.1.8 Оборудование для нанесения образцов
Торрированные стеклянные капилляры
Ассортимент капилляров: 1, 2, 3, 5, 10, 20 мкл. Использование держателя капилляров "Микрокап" позволяет достигать высокой точности дозировки вручную.
100 штук капилляров комплектуется 1 шт. "Микрокап".
Шприцевой дозатор "MULTISTEP-50" в комплекте с микрошприцем V=1 мкл (Аналог РВ-600 фирмы Hamilton)
Шприцевой дозатор "MULTISTEP-50" предназначен для многократной дозировки микроколичеств жидкости с помощью микрошприцев. "MULTISTEP-50" обеспечивает пятидесятикратное повторение дозы (50 шагов). Для микрошприцев с рабочим объемом 1 мкл каждый шаг дозатора "MULTISTEP-50" отмерит 0,02 мкл жидкости. Дозатор "MULTISTEP-50" существенно облегчает работы, связанные с многократным повтором микродоз.
Столик для нанесения образцов на пластины (с подогревом)
Использование для нанесения проб на пластины столика с подогревом дает возможность получения пятен стартовых зон минимального размера (2-3 мм), что необходимо для более эффективного разделения многокомпонентных смесей при хроматографии.
3.1.9 Приборы и оборудование для высокоэффективной тонкослойной хроматографии
СИСТЕМА ДЛЯ ТОНКОСЛОЙНОЙ ХРОМАТОГРАФИИ С ДЕНСИТОМЕТРОМ "ДенСкан"
Назначение и область применения
Системы для тонкослойной хроматографии и электрофореза с денситометром "ДенСкан" предназначены для качественного и количественного анализа состава проб веществ и материалов в видимой области спектра и ультрафиолетовом свете при длинах волн 254 и 365 нм. Область применения - исследования в химии, биохимии, биологии, медицине, фармакологии, аналитическом контроле чистых веществ, объектов окружающей среды и др.
Технические данные
· Денситометр обеспечивает расчет параметров и количественную оценку хроматограмм в видимой и ультрафиолетовой области спектра (max= 254 нм, max =365 нм)
· Размер обрабатываемых пластин, не более 15 х 15
· Время ввода изображения, с не более 5
· Время обмера хроматограммы, мин 5
· Отношение сигнал/шум: видимая область не менее 5/1УФ, 254 нм не менее 5/1УФ, 365 нм не менее 5/1
· Относительное СКО по площади пятен, % видимая область не более 5УФ, 254 нм не более 5УФ, 365 нм не более 5
· Размах значений Rf : видимая область не более 0,02УФ, 254 нм не более 0,02УФ, 365 нм не более 0.02
· Масса осветительной камеры, кг не более 12 кг
· Габаритные размеры осветительной камеры, мм не более длина. 420 ширина 420 высота 700
· Напряжение питания, В 220 ± 22/33
· Частота переменного тока, Гц 50 ± 1
· Средняя наработка на отказ денситометра, ч не менее 5000
1. Состав денситометра
2. Денситометр "ДенСкан" состоит из камеры осветительной, черно-белой либо цветной видеокамеры или сканнера, блока ввода изображения, системы обработки данных. Камера осветительная выполнена в виде блочной конструкции, включающей следующие основные узлы:- источники света: лампы дневного света. Лампы УФ диапазона, длина волны 254 нм лампы УФ диапазона, длина волны 365 нм- набор корректирующих светофильтров- детектор - черно-белая малогабаритная видеокамера OS-45D или аналогичная с чувствительностью не хуже 0,02 люкс, с ручной фокусировкой и ручной регулировкой диафрагмы либо цветной сканнер с разрешением от 200 d.p.i. и выше с интерфейсом, соответствующим TWAIN стандарту- установочный столик для пластин; - канал связи с блоком ввода изображенияСистема обработки данных с использованием персонального компьютера и программного обеспечения "Dens". Минимальные требования к компьютеру:- Операционная система - Microsoft Windows 95, Windows 98, Windows NT (версия 4.0 или выше) - Процессор - Pentium 100 MHz- Оперативная память (RAM) - 8 Мбайт, рекомендуется 16 Мбайт- Цветной монитор - с диагональю не менее 14 дюймов- Место на жестком диске - 10 Мбайт- Манипулятор - "мышь"Блок ввода изображения видеобластер AverMedia ( и программное обеспечение к нему) используется для получения изображения хроматограммы на мониторе компьютера. Возможно использование аналогичных систем. [12]
Литература
1. Аналитическая хроматография / Под ред. В.И. Сакодынского и др. М.: Химия, 1993, с. 19 – 21.
2. Кнорре Д.Г., Крылова Л.Ф., Музыкантов В.С. Физическая химия. - М.: Высш. шк., 1990. - 416с.
3. Алесковский В.Б., Бардин В.В., Булатов М.И. Физико-химические методы анализа. Практическое руководство.- Л.: Химия, 1988. – 376 с.
4. Ольшанова К.М. Практикум по хроматографическому анализу. М., Высш. школа, 1970. -312с.
5. Определение ддт, ддэ, ддд, альдрина, дильдрина, гептахлора, кельтана, метоксихлора, эфирсульфоната и других ядохимикатов в продуктах питания хроматографией в тонком слое / Методы определения микроколичеств пестицидов в продуктах питания, кормах и внешней среде. М.: Колос, 1977. с. 9 – 17
6. Е.С. Косматый, Б.М. Тверская, Ф.И. Полонская. Определение о,о-диэтил-8-(6-хлорбензоксазолинил-3-метил)-дитиофосфата (фозалона) в яблоках методом тонкослойной хроматографии / Методы анализа пестицидов // Проблемы аналитической химии. Том II / М., «Наука», 1972. С. 70 – 73.
7. W.Mc Kinley, S.J. Grahem. J. Assoc. Offic. Agric. Chemists, 43, 89 (I960).
8. L. Fishbein, J. Fowkes, P. Jones. J. Chromatogr., 23, 47b (1966).
9. Р. Nangniot, G. Dardenne. Bull. Inst. Agr. St. Res., 31, 120 (1963).
10. A.R. Kittleson. Analyt. Chem., 24, 1173 (1952).
11. Л.М. Фукельман. Сборник трудов ВИЗР, вып. 20, ч. 4, 61 (1964).
12. http://lenchrom.spb.ru/chromatography/thinlayer_004.shtml
Перепечатка материалов без ссылки на наш сайт запрещена